This page describes the usage of the command line interface of KOGAL. The executable KOGAL is compiled for Linux x86_64 and Windows 64bit platform. The KOGAL python source code and the datasets used to generate such results are publicly accessible online https://perso.isima.fr/~enmephun/FILES/KOGAL/KOGAL.zip
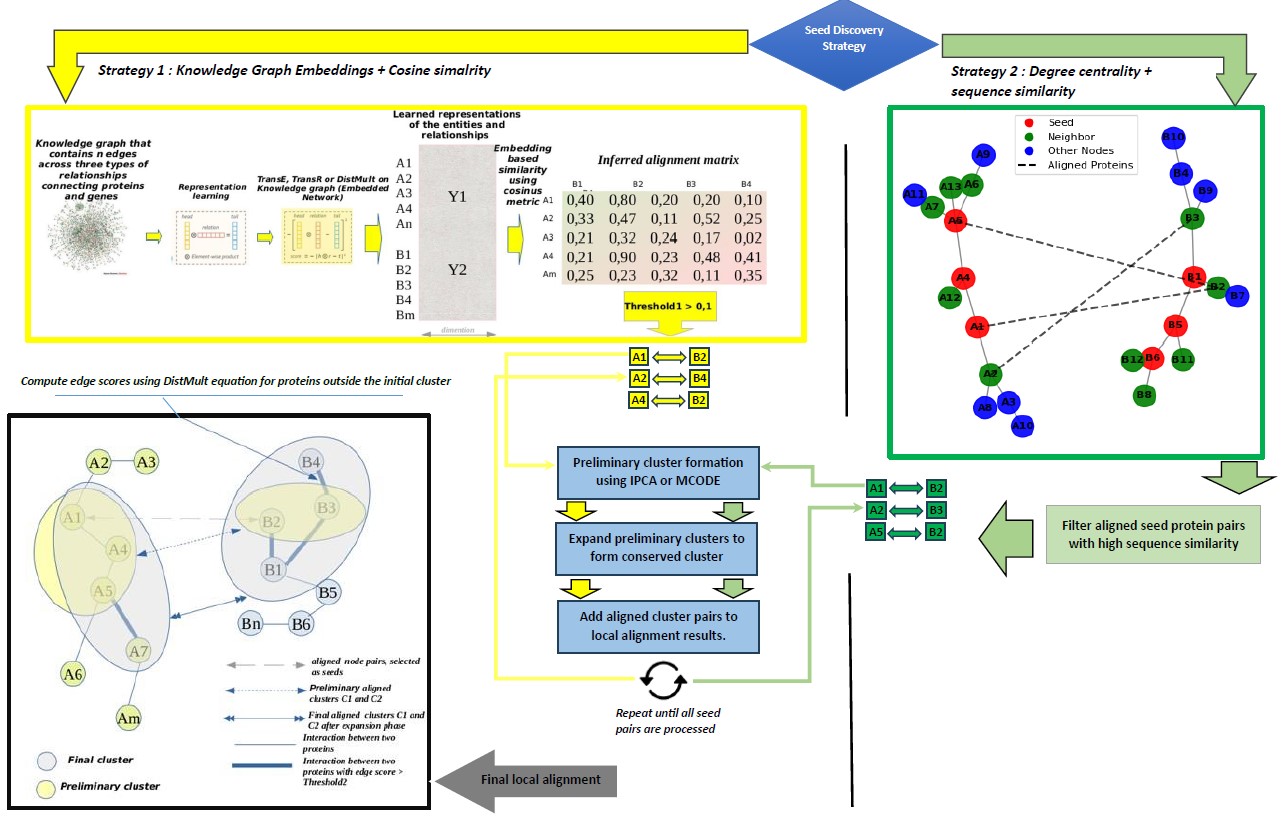
KOGAL aims to predict conserved protein complexes across species’ PPI networks. It employs two strategies for seed discovery and initial alignment: the first computes the cosine similarity between embedding vectors derived from knowledge graph models like TransE or DistMult to generate an alignment matrix. The second strategy enhances alignment by calculating the degree of centrality of nodes within each network, capturing the importance of each protein in the network structure. Protein similarities are quantified by combining protein sequence similarities with knowledge graph embeddings, ensuring biologically meaningful structural alignments.
python3 KOGAL.py --clstm ipca
python3 KOGAL.py --clstm mcode
usage: KOGAL.py [] [-h | --help] Local network alignment using Knowledge Graph Embedding Models options: -h, --help show this help message and exit --policy.txt Paths for BLAST files and networks --strategy (strategy == 1) the alignment process starts by computes the cosine similarity between embedding vectors derived from knowledge graph models. (strategy == 2) the alignment process starts by calculating the centrality degree of nodes within each network, highlighting the importance of each protein in the network structure. -- N The top N proteins with the highest centrality are selected as seeds --SEED_DC Applying a threshold to filter the pertinent seed node pairs of (strategy ==1) in order to detect the pairs of initial clusters --entity_emb_path Entity embedding file --relation_emb_path Relation embedding file --entity_idmap_path Entity mapping file --relation_idmap_path Relation mapping file --gamma GAMMA --SCORE_THRESHOLD SCORE_THRESHOLD Minimum score needed for cluster detection. Any cluster whose score is less than a given threshold is abandoned --SEED_THRESHOLD SEED_THRESHOLD Applying a threshold to filter the pertinent seed node pairs in order to detect the pairs of initial clusters --alpha ALPHA Tuning the contribution between the local and global edge score computed from the knowledge graph embedding -save SAVE_PATH, --save_path SAVE_PATH --clstm CLSTM Choosing graph clustering techniques (i.e. ipca, mocode or coach)